Gaucher Disease
Gaucher disease is an autosomal recessive genetic disease and the most common lysosomal storage disease with a frequency of approximately 1 in 50,000 habitants in Central Europe. It is caused by a lack of a specific enzyme (glucocerebrosidase) in the body, which in turn is caused by a genetic mutation inherited from both parents (autosomal recessive inheritance). The lack of glucocerebrosidase entails the accumulation of the substrate glucocerebroside which accumulates in the spleen, liver, kidneys, lungs, brain and bone marrow. The courses of the disease may differ and vary from no external symptoms to severe disability and death.
The symptoms include enlarged spleen and liver, liver dysfunction, bone diseases or painful bone defects, neurological complications, swelling of the lymph nodes and (occasionally) joint defects, bloated abdomen, brownish skin complexion, anemia, low blood platelet count and yellow fat deposits on the sclera of the eye. The most severely affected can also be more susceptible to infections. Despite the fact that Gaucher disease shows a phenotype with different degrees of severity, it was divided into three subtypes depending on the presence or absence of neurological involvement.
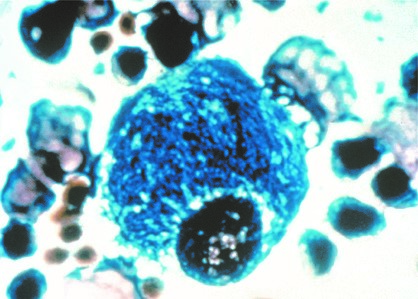
Gaucher disease is referred to as „lipid storage disease“, in which abnormal amounts of lipids, so-called „glycosphingolipids“, accumulate in special reticuloendothelial cells. In classic cases, the cell nucleus is pushed to the edge of the cell and the cell is filled with abnormal lipids.
Types of Gaucher Disease
Typ I, the most common type, also called ’non-neuropathic‘ type, is characterized by the problems mentioned above. It is often referred to as an adult form, although its cause is already existent at conception. The median age at diagnosis is 28 years and life expectancy is slightly reduced. In most cases there are no major neurological symptoms.
Typ II, also called acute neuronopathic Gaucher disease, occurs rarely and is characterized by rapidly progressing neurological problems in newborns. Formerly referred to as infantile Gaucher disease, type II is characterized by severe neurological involvement during the first year of life. Fewer than 1 in 100,000 newborns suffer from type 2. The prognosis is grim: because of the heavy involvement of the nervous system, an affected child rarely exceeds the age of 2 years.
Typ III, also called neuronopathic Gaucher disease, is also very rare, but in contrast to type II, it is characterized by slowly progressing neurological symptoms. The signs and symptoms of type III Gaucher disease appear later in childhood than the symptoms of type II.
